
为什么有了临床试验,器械还是进不了医院?
医疗器械的创新往往从一个看似简单的愿望开始:让手术更安全,让诊断更精准,让患者恢复更快。然而,真正能进入医院日常、写进指南路径的器械,却屈指可数。
这背后最大的障碍之一,就是证据。
很多企业有过这样的困惑:产品已经完成了注册临床试验,甚至拿到上市许可,但医生依旧迟疑,医保迟迟不纳入,市场始终打不开。为什么?因为“做过临床试验”不等于“证据充足”。
数据显示,美国 FDA 每年平均批准2929个 510(k) 器械,但真正代表创新原理、必须依赖系统临床证据的 PMA 审批数量只有约34个 。进入临床路径的比例更低。换句话说,只有少部分器械走到了“证据站得住”的那一步。
证据不仅是审批环节的一张“入场券”,它还同时承担着三重角色:让监管机构相信安全有效,让医生愿意在临床实践中使用,让支付方认可其经济与社会价值。临床证据,是器械真正走向常规应用的“通行证”。
01
临床证据的多重角色
审批的通行证
在全球主要市场,临床证据是注册审批的核心环节。
中国:NMPA 明确规定,对新原理、新结构的创新器械,必须开展系统临床试验,提交安全性与有效性数据。如果有充分的临床评价资料,也可申请豁免,但比例有限。
美国:FDA 对高风险(Class III)器械要求通过 IDE(Investigational Device Exemption) 临床试验,才能支撑 PMA(Pre-Market Approval) 申请 。
欧洲:随着 MDR 上线,临床证据要求显著提高。过去部分器械可以通过“等效性”证明完成注册,如今必须提交自身的临床数据,对创新产品尤其严格 。
审批层面的证据要求,确保了器械“能安全有效地上市”。但这只是第一道门槛。
医生采用的基础
审批通过,并不意味着医生会立即把新器械纳入日常工作。医生真正看重的,是证据是否能回答:
它比现有技术更安全吗?
它能显著改善患者结局吗?
它的并发症、长期随访数据是否可靠?
可吸收冠脉支架就是一个典型案例。早期的临床试验结果显示手术成功率和短期效果良好,产品也获得了批准。但随后多年随访发现,再狭窄和支架塌陷等并发症明显高于金属支架,导致医生群体对其信任度急剧下降,临床采用率随之下滑。
这说明:医生的采纳,依赖的是长期、扎实的证据,而不仅仅是审批文件里的数据。
支付与市场进入的依据
对支付方而言,证据还承担着第三重角色:决定是否为创新买单。
欧美:健康技术评估(HTA)几乎是支付决策的必经环节。英国 NICE 的 HTA 手册明确要求,必须在比较不同干预手段的基础上,结合经济建模评估器械的“成本-效果比” 。没有强有力的经济学证据,新技术很难进入医保或国家健康体系。
美国:2024 年,CMS 发布了 TCET(Transitional Coverage for Emerging Technologies) 最终通知,允许部分创新设备在有限覆盖的同时,继续积累真实世界证据(Coverage with Evidence Development, CED) 。这体现出证据对支付环节的“边覆盖、边验证”价值。
中国:近年来,国家医保局逐渐引入临床和经济学评估思路,在创新耗材准入、集采之外的谈判环节,越来越强调“真实临床价值”。
换句话说,即使一款器械拥有监管许可,如果不能用证据证明它不仅“能用”,还“值得用”,支付体系也不会轻易放行。
02
为什么“试验结果”还不够?
很多企业会问:我的产品已经做了临床试验,为什么还是没能顺利进入医院常规?原因在于,监管审批所需的证据,与医生采用和支付决策所需的证据,并不是一回事。
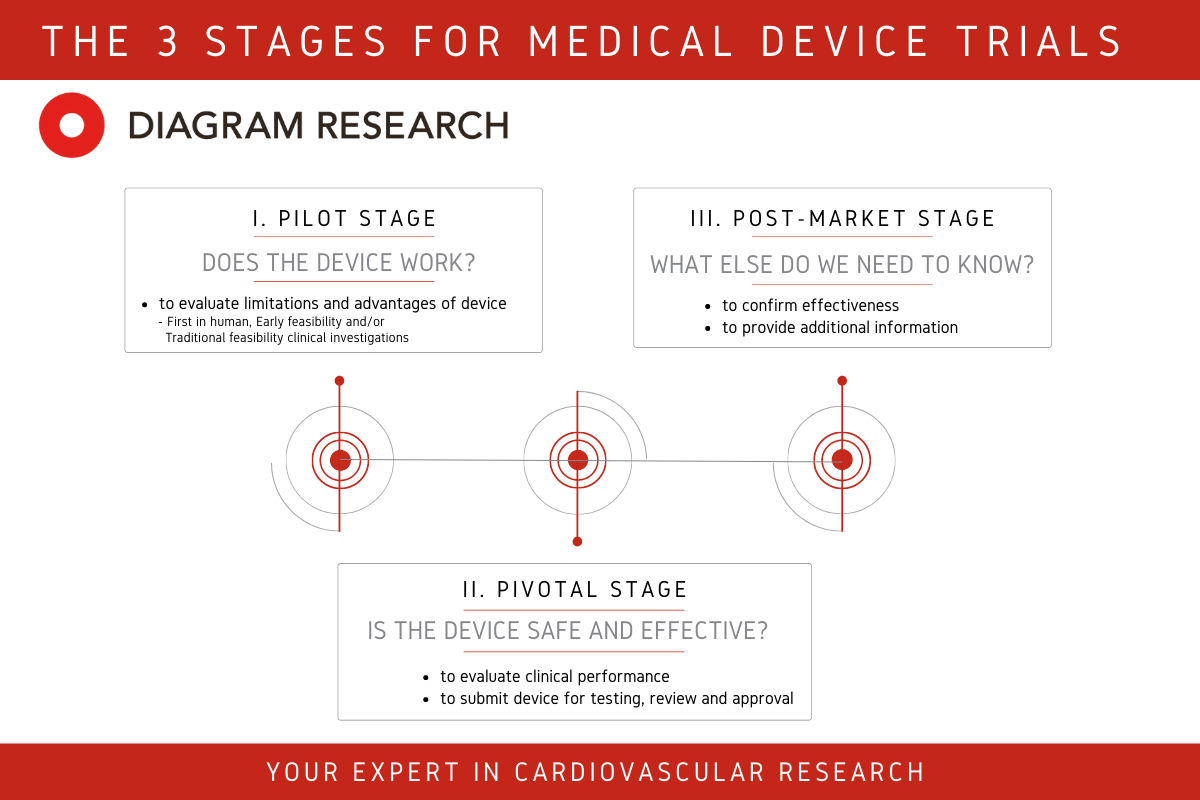
小样本与外部效度不足
不少注册临床试验规模有限,常见设计是单中心、数十至数百例。这样的数据能够回答“能不能用”,但无法回答“普遍能不能用”。欧洲 MDR 明确要求,临床评价必须基于“足够的样本量与代表性”,强调外部效度。因此,过去依赖“等效性”豁免的路径越来越难走,新产品必须自己积累更多、多中心的证据 。
随访周期过短
医疗器械,尤其是植介入和耐用设备,往往需要多年随访才能看清长期风险。以可吸收支架为例:上市时的试验主要观察 1 年内的再狭窄率,结果良好。但 3–5 年随访显示,其再狭窄和塌陷并发症风险显著高于金属支架,最终导致市场信心丧失。这说明,短期数据可以过审,但不足以赢得医生的长期信任。
终点指标不匹配
企业在试验中常设“过程指标”——例如手术时间缩短、影像指标改善。但医生和支付方更关心的是临床结局(并发症率、再手术率、功能改善)和经济性(是否能降低总体医疗开支)。
英国 NICE 在其 HTA 评估手册中,要求评价必须涵盖“目标人群、比较方案、主要结局指标以及成本-效果建模” 。这意味着:如果企业的试验终点没有覆盖患者获益和经济价值,再漂亮的数据也难以推动支付方放行。
器械迭代过快,传统 RCT 不完全适用
软件类医疗器械和 AI 影像产品更新频繁,往往一年就能迭代多个版本。传统 RCT 从设计到完成可能要花三五年,结果出来时,产品版本已更新,证据“过期”。
因此,监管机构开始转向接受真实世界证据(RWE)作为补充。FDA 在 2023 年更新的 RWE 指南草案里提出,可以在满足数据质量和偏倚控制的前提下,将真实世界数据用于监管决策 。这为快速迭代的器械提供了新的证据路径。
资金与资源的现实制约
做一项多中心、长期随访的器械临床试验,往往需要数千万乃至上亿元投入。对初创企业来说,难度极高。美国 NIH 等机构设有“转化基金”来支持,但覆盖有限;中国也在探索通过乐城真实世界研究试点来帮助企业补证 。这也解释了为什么很多项目停留在“能过审批”,却无法积累更高层级的证据。
03
真实世界证据(RWE)的崛起
当传统随机对照试验(RCT)在样本量、周期和适应场景上显得捉襟见肘时,真实世界证据(RWE)正逐渐走上舞台。它并不是替代 RCT,而是对器械证据链的有力补充。
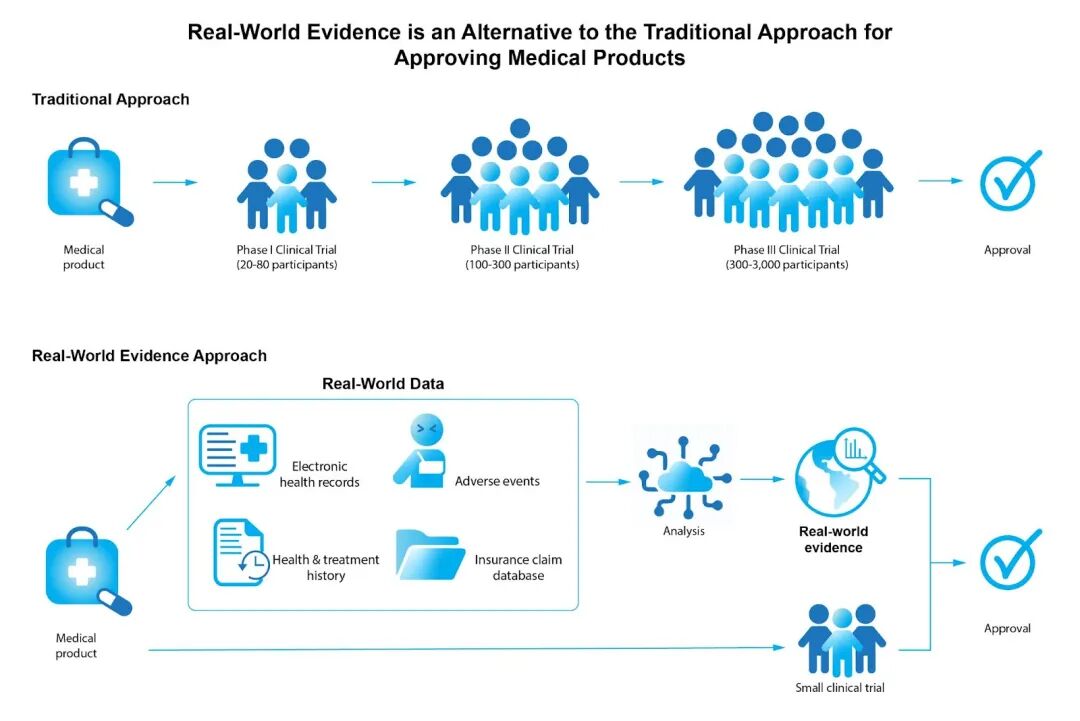
监管侧:从观望到制度化
美国 FDA:早在 2016 年就提出将 RWE 纳入监管决策的框架,并在 2023 年发布了更新的指南草案,明确要求先评估数据来源、质量和偏倚,再决定是否用于审批 。目前,AI 影像软件、植介入器械更新迭代等场景,FDA 已经多次引用 RWE 支撑决策。
欧盟 MDR:要求企业在提交临床评价报告(CER)时,必须纳入上市后临床随访(PMCF)的真实世界数据,作为长期风险和性能验证的核心。
中国 NMPA:2019 年起在海南乐城率先启动真实世界数据(RWD)试点,允许企业用境外上市器械在乐城产生的临床使用数据,作为全国注册申报的补充证据 。截至 2023 年,已有 20+ 款器械通过该路径进入中国市场。
这意味着,RWE 已经从“尝试性探索”变成了制度化的一环。
临床侧:补足证据的盲区
对医生而言,RCT 的人群筛选往往过于理想化:排除了多病共存、高龄患者,而这些恰恰是现实临床的主要人群。RWE 能够在大样本、多中心、日常实践场景下,验证器械在“真实患者”中的表现。
例如,经导管主动脉瓣置换术(TAVR)最初的试验人群局限于高危患者,但随着大量 RWE 的积累,证明其在中低危患者中同样有效且安全,最终推动了适应症扩展。没有真实世界数据,这一步几乎不可能完成。
支付侧:边覆盖、边补证
RWE 的另一个重要舞台,是支付。
美国 CMS:2024 年发布 TCET(Transitional Coverage for Emerging Technologies) 最终方案,明确允许部分突破性器械在有限覆盖的同时,继续生成真实世界证据(Coverage with Evidence Development, CED) 。这让企业能够“先进入医保”,同时补足长期证据,避免了在证据不足时被完全挡在支付体系之外。
中国医保:虽然尚未像 CMS 那样制度化,但在部分地方性医保谈判和高值耗材准入中,越来越强调“真实临床数据”的价值。乐城 RWE 的成功经验,也为支付端探索提供了先例。
企业视角:新的竞争点
RWE 的兴起,意味着证据生产不再是“一次性任务”,而是一个全生命周期过程。企业不仅要在上市前做试验,还要在上市后持续追踪、积累数据。谁能把临床随访、注册系统、真实世界研究做成标准化能力,谁就更有可能获得医生和支付方的长期信任。
04
中外证据体系差异
医疗器械的证据体系,在不同国家和地区呈现出鲜明差异。这种差异,既反映了监管环境的不同,也折射出产业阶段和支付体系的差别。
欧美:循证为先,证据与支付紧密挂钩
在美国和欧洲,临床证据不仅是审批要求,更是支付与指南纳入的前置条件。
美国:FDA 审批强调安全性与有效性,而一旦进入临床路径,还需要通过 HTA(健康技术评估) 与医保覆盖评审。NICE 在英国就是典型代表,评估框架明确要求比较多方案、结合经济学建模,最后决定是否推荐纳入 NHS 。
CMS 的 TCET 更是一个标志性转变:对突破性器械允许“有限覆盖”,同时要求企业持续生成真实世界证据 。换句话说,证据的作用不仅限于上市,而是覆盖了审批—支付—应用的完整链条。
欧美体系的优势在于严谨,劣势在于周期长、投入大。数据显示,一个典型的高风险器械,从首批试验到进入支付体系,往往需要 5—10 年的证据积累。
中国:审批提速,证据体系正在补课
中国的医疗器械创新过去十年快速崛起,但在证据体系上,路径仍在探索:
审批层面:NMPA 的“创新医疗器械特别审评”通道,让不少产品在研发初期就获得了监管部门的沟通与加速,显著缩短了上市周期。
真实世界试点:海南乐城的 RWE 政策是中国的制度创新,为境外器械快速补证、加速国内上市提供了范例 。
支付端:虽然国家医保局已在谈判和集采环节强调“临床价值”,但尚未形成如 NICE 一样系统的 HTA 框架。更多时候,证据和支付的对接依赖于试点和个案。
这意味着,中国的器械转化往往能“更快获批”,但在走向医生广泛采纳和医保全面覆盖时,仍需补齐“高质量证据链”。
共性规律:证据是核心竞争力
无论在欧美还是中国,那些最终能走向常规应用的器械,都有一个共同点:证据链条完整。
审批证据:保证安全有效;
临床证据:获得医生认可;
经济学证据:赢得支付支持;
长期证据:通过随访和真实世界研究,不断巩固信任。
差别只是在于:欧美是制度化要求,中国是在逐步完善。但方向是一致的——没有证据,再好的技术也只能停留在“上市”而不是“应用”。
05
结语:证据即通行证
在医疗器械的转化过程中,证据从来都不是一份“附属文件”,而是决定技术能否真正走到患者身边的核心。
审批层面的临床试验,可以让一款产品“获得身份”;医生认可的长期证据,才能让它“进入手术室”;支付体系要求的经济学证据,才能让它“进入市场常规”。
在欧美,这一逻辑已经制度化:RCT + RWE + HTA 构成了完整的证据生态。产品要想进入指南和医保,必须一步步补齐链条。过程漫长,但成功后,器械往往能稳定存在多年。在中国,创新器械的上市速度越来越快,但能否持续落地,还取决于证据体系的建设。从乐城 RWE 试点,到医保谈判对“真实临床价值”的强调,都在说明:中国正在把证据从“审批必需品”转向“应用核心力”。
这也意味着:未来的竞争,不仅仅是技术的比拼,更是证据生产能力的较量。谁能更快、更系统地产生高质量的临床与真实世界证据,谁就更有机会获得医生的信任、支付方的支持和市场的认可。
对于医生来说,证据是判断一款器械是否值得使用的依据;对于企业来说,证据是打开市场的护照;对于投资人来说,证据是创新能否变现的试金石。
证据不是终点,而是创新真正的起点。
参考链接:
1.U.S. Food & Drug Administration (FDA). Investigational Device Exemption (IDE).
2.U.S. Food & Drug Administration (FDA). Breakthrough Devices Program & RWE Guidance (2023 draft).
3.European Commission. Medical Device Regulation (MDR) Clinical Evaluation Requirements.
4.National Institute for Health and Care Excellence (NICE). Health Technology Assessment Manual.
5.Centers for Medicare & Medicaid Services (CMS). Transitional Coverage for Emerging Technologies (TCET), 2024 Final Notice.
6.National Medical Products Administration (NMPA). 创新医疗器械特别审评审批程序. 国家药品监督管理局,2014;海南乐城 RWE 试点政策,2019
